Research
Software Development and Tools Colloidal materials Biological membranes
Software Development and Tools
|
|
|
|
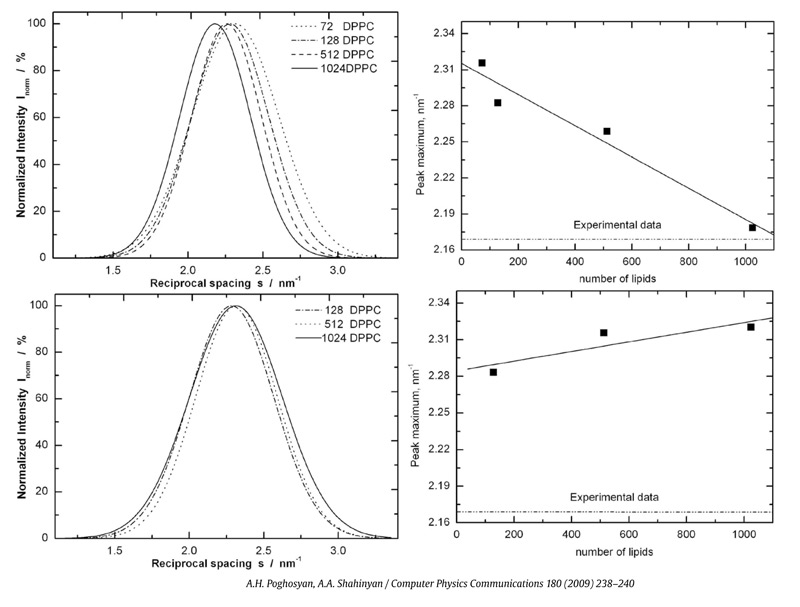
 Software benchmarking and comparison: It is reasonable to divide existing software packages into two major categories, i.e. computational and constructional. Usually, the computational software packages include molecular mechanics using various force fields, like GROMACS, NAMD, AMBER, etc, semi-empirical and ab initio quantum mechanical calculations, such as GAMESS , MOPAC, GAUSSIAN, etc. The constructional software packages are mostly designed for molecule or system constructions, visualization and some drawing tools, being as multi-purpose packages (VDM, RasMol, Hyperchem, etc). For our MD simulations, we use NAMD and GROMACS with open source codes, which are aimed at the high performance simulation with parallel support. Recently, the comparison of NAMD and GROMACS has been done by us, where comparable feature analysis of both packages has been carried out. It was stated that the GROMACS has been displayed as faster as NAMD, which is probably due to united atom character, meanwhile NAMD is more suitable for simulation of relatively small systems and for detailed analysis of the system in all atom character. It was also established that NAMD shows linear increase with increase of number of processors, however GROMACS receives saturation and even goes to the worst results. The parallel scaling (up to 48 cores) of NAMD package has been studied by estimation of the sensitivity of interconnection on speedup and benchmark results – testing the parallel performance of Myrinet, Infiniband and Gigabit Ethernet based networks, where the systems with various sizes have been used (1000K, 330K, 210K, 110K, 92K, 54K, 27K and 16K). The Armenian grid infrastructure – ArmGrid has been used as a main platform for series of benchmarks. According to the benchmarking results, due to the high performance of Myrinet and Infiniband networks, the Myrinet based cluster and the cluster located in theYerevanStateUniversity (Infiniband network) show reasonable values, meanwhile the scaling of clusters with various types of Gigabit Ethernet interconnections breaks down when interconnection is activated. However, the clusters equipped by Gigabit Ethernet network are sensitive to change of system, particularly for 1000K systems no breakdown in scaling was reached. The Infiniband equipped cluster in comparison with Myrinet, makes it possible to receive almost ideally results regardless of system size. We have also suggested a benchmarking formula, which provides the computational throughput depending on the number of processors. To check the benchmarking formula, we have also performed benchmark tests of 210K system on Blue Gene/P supermachine (IBM Blue Gene/P: PowerPC 450 processors, a total of 8192 cores) at Bulgarian Supercomputing Centre. In overall, we find the good agreement with estimated findings. These results should be important, for instance, to choose most appropriate amount of processors for studied system. The benchmarking on GROMACS package is in progress. Software benchmarking and comparison: It is reasonable to divide existing software packages into two major categories, i.e. computational and constructional. Usually, the computational software packages include molecular mechanics using various force fields, like GROMACS, NAMD, AMBER, etc, semi-empirical and ab initio quantum mechanical calculations, such as GAMESS , MOPAC, GAUSSIAN, etc. The constructional software packages are mostly designed for molecule or system constructions, visualization and some drawing tools, being as multi-purpose packages (VDM, RasMol, Hyperchem, etc). For our MD simulations, we use NAMD and GROMACS with open source codes, which are aimed at the high performance simulation with parallel support. Recently, the comparison of NAMD and GROMACS has been done by us, where comparable feature analysis of both packages has been carried out. It was stated that the GROMACS has been displayed as faster as NAMD, which is probably due to united atom character, meanwhile NAMD is more suitable for simulation of relatively small systems and for detailed analysis of the system in all atom character. It was also established that NAMD shows linear increase with increase of number of processors, however GROMACS receives saturation and even goes to the worst results. The parallel scaling (up to 48 cores) of NAMD package has been studied by estimation of the sensitivity of interconnection on speedup and benchmark results – testing the parallel performance of Myrinet, Infiniband and Gigabit Ethernet based networks, where the systems with various sizes have been used (1000K, 330K, 210K, 110K, 92K, 54K, 27K and 16K). The Armenian grid infrastructure – ArmGrid has been used as a main platform for series of benchmarks. According to the benchmarking results, due to the high performance of Myrinet and Infiniband networks, the Myrinet based cluster and the cluster located in theYerevanStateUniversity (Infiniband network) show reasonable values, meanwhile the scaling of clusters with various types of Gigabit Ethernet interconnections breaks down when interconnection is activated. However, the clusters equipped by Gigabit Ethernet network are sensitive to change of system, particularly for 1000K systems no breakdown in scaling was reached. The Infiniband equipped cluster in comparison with Myrinet, makes it possible to receive almost ideally results regardless of system size. We have also suggested a benchmarking formula, which provides the computational throughput depending on the number of processors. To check the benchmarking formula, we have also performed benchmark tests of 210K system on Blue Gene/P supermachine (IBM Blue Gene/P: PowerPC 450 processors, a total of 8192 cores) at Bulgarian Supercomputing Centre. In overall, we find the good agreement with estimated findings. These results should be important, for instance, to choose most appropriate amount of processors for studied system. The benchmarking on GROMACS package is in progress. |
Colloidal materials
|
Surfactant bilayers: Surfactants, i.e. molecules consisting of a polar head and a long alkyl tail, generate quite different self-assembled structures, from micelles to lyotropic liquid crystalline ones. The later is composed of individual lamellae of fluid amphiphilic molecules separated by a solvent. Surfactant bilayers have been intensively studied for many years, and can be considered as a simple model for biological membranes. It should be noted that surfactants have been found already a wide range of applications in everyday life, in detergents, cosmetics, pharmaceuticals, food processing, agrochemicals, paints, paper coatings, etc. By using well-known ionic surfactant molecules sodium dodecyl sulfate (SDS) and sodium pentadecyl sulfonate (SPDS), we have investigated the lyotropic liquid crystalline phases of both molecules. A series of long 130ns molecular dynamics simulations of SDS/water system (512 SDS/15.000 water) were done using all atom and the united atom models. As software we use both NAMD and GROMACS codes with CHARMM27 and modified GROMOS87 force fields, respectively. It has been stated that the CHARMM force field with all atom model gave the best results in comparison to the experimental findings, i.e. we found a strong agreement with the experimental values of area per molecule and hydrocarbon tail packing parameters. The above mentioned job was jointly done with our partners from the Institut für Chemie (Universität Potsdam) inPotsdam. The next step was the detailed study of long chain alkyl sulfonate/water system in lamellar phase, where as a surfactant SPDS molecule was used. In this regards, about 600ns parallel molecular dynamics simulation study was conducted for SPDS/water system. As the starting configuration was random, we examined the mechanism of self-assembly of SPDS molecules, i.e. we have explored in detail the dynamics of bilayer formation, which is indeed quite interesting as related to the dynamic self-assembly pathway leading to lamellar phase. Another interesting phenomenon was archived using the simulated annealing treatment. The temperature changing leads to the formation from the gel to fluid like phase with fully disordered hydrocarbon chains, i.e. the system undergoes gel-to-fluid phase transition. |
|
||||||||||
|
|
|||||||||||
|
Inverse micelle/polyelectrolyte complexes: In collaboration with the Institut für Chemie (Universität Potsdam) in Potsdam, we have performed MD experiments of inverse sodium dodecyl sulfate (SDS) micelles in a mixed toluene/pentanol solvent in the absence and presence of a cationic polyelectrolyte, i.e. poly (diallyldimethylammonium chloride) (PDADMAC) aimed to estimate the influence of the PDADMAC on structural parameters of the microemulsion, such as the water droplet size, water properties or viscosity. The receiving MD data indicate a more rigid and ordered surfactant film due to the formation of a polyelectrolyte palisade layer in full agreement with the experimental findings, e.g. the viscosity increase and shift of the percolation boundary. |
|
||||||||||
|
Surfactant bilayer/polyelectrolyte complexes: We have carried out a 50 ns of molecular dynamics study of PDADMAC/SDS/decanol/water systems, where the influence of the cationic polyelectrolyte on the anionic SDS-based lamellar liquid crystalline system was investigated. The polyelectrolyte-induced coexistence of two lamellar phases at a concentration of 2-3% of PDADMAC was observed, which is in agreement with experimental findings. The obtained MD results also provide us important information on the dynamical and structural features of the abovementioned complex system, as well as the conformational properties of the polyelectrolyte and the surfactant molecules become available. This work also was done in collaboration with the Institut für Chemie (Universität Potsdam) in Potsdam.
|
|
||||||||||
|
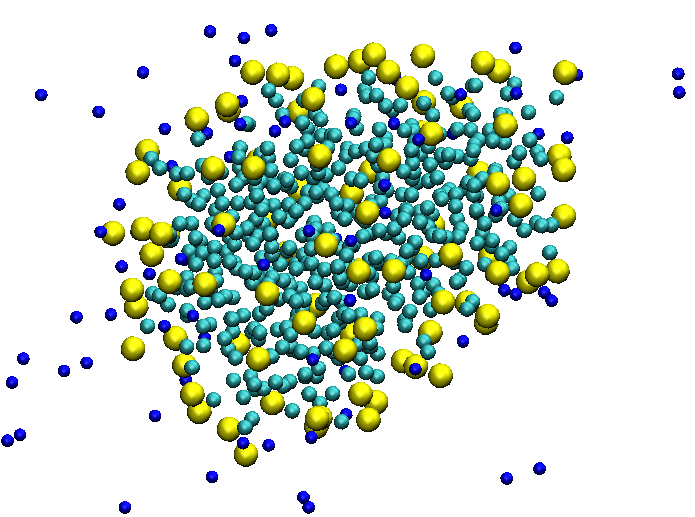
Coarse-grained simulations: The coarse-grained (CG) approach, which is originally developed by Marrink and coworkers, provides to simulate huge systems with long time periods. We tried to study the mechanism of the self-assembly of long alkyl tail ionic micelles using the so called “granular” approach. During last decade, a number of simulations were reported, where many authors claim that it is very difficult to reach an equilibrium using atomic scale MD, as it is known that the lifetime of micelle formation is an order of microseconds. In this regards, we have carried out CG simulations using GROMACS code with MARTINI force field. We have reached up to 80μs timescale and tracked final formation at 50μs timepoint. A key structural parameters, such as radius of gyration, characteristic relations of chain were estimated to understand the mechanism of self-organization process.
|
 |
||||||||||
|
Biological membranes
|
|||||||||||









